

Abstract
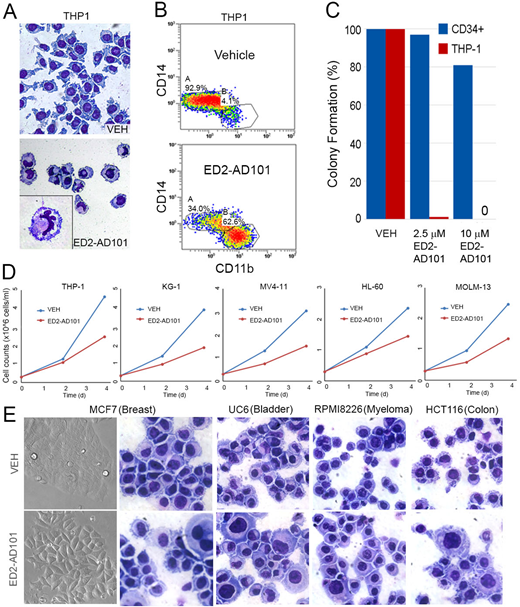
Acute myeloid leukemia (AML) cells express the PU.1/RUNX1/CEBPA master transcription factor circuit at levels similar to or exceeding that in normal granulocyte-monocyte progenitors (GMP), but somehow terminal granulo-monocytic fates do not occur. Gene expression analyses of AML cells compared to the normal hematopoietic hierarchy confirmed suppression of hundreds of monocyte and granulocyte terminal-differentiation genes that were PU.1 targets by chromatin-immunoprecipitation analyses, despite intact expression of proliferation and myeloid-commitment genes. To better understand this repression rather than activation by the PU.1/RUNX1/CEBPA circuit, we used liquid chromatography-tandem mass spectrometry (LCMS/MS) to analyze the protein composition of the PU.1/RUNX1/CEBPA master transcription factor hub in AML cells of various genotypes. A striking common finding was enrichment for corepressors, coregulators that repress gene transcription, over coactivators that activate genes (we have previously described how leukemia oncoproteins produce this corepressor/coactivator imbalance [e.g., Gu et al., JCI 2018]). Prominent amongst the enriched corepressors were ATP-dependent chromatin remodelers SMARCA5 and CHD4 that execute the critical and energetically expensive event in epigenetic repression of nucleosome positioning to obstruct gene transcription start sites. To translate these observations towards therapy, we used a high throughput screen to identify a first-in-class compound series (ED2-AD101) that inhibits SMARCA5/CHD4. In silico modeling, supported by structure-activity relationship (SAR) studies, surface plasmon resonance and ATP-ase assays, indicated that ED2-AD101 inhibits ISWI family ATP-ase activity allosterically by binding to the highly conserved HELICc-DExx domain. In cell-based assays, ED2-AD101 at doses of between 1 to 10 µM potently suppressed AML cell growth (THP1, KG-1, MV411, HL60, MOLM13) via terminal-differentiation (without early apoptosis) while simultaneously sparing exponential growth of normal CD34+ hematopoietic stem and progenitor cells (Fig.1 A, C, D). We also demonstrated terminal granulo-monocytic differentiation using CD11b/CD14 markers by flow cytometry in THP-1 cells treated with ED2-AD101 at 1 µM dose (Fig 1B). Importantly, these differentiation-based cell cycle exits occurred even in chemo-resistant p53-null AML and other cancer cells (MCF7, UC6, RPMI 8226, and HCT116 cancer cells treated with 10µM of ED2-AD101. In the first panel of Fig. 1E, MCF7 cells show morphologic changes indicative of epithelial differentiation after treatment. Giemsa-stained cytospin preparations of all the above cell lines displayed morphological features of cell differentiation, such as a lower nucleocytoplasmic ratio after treatment) (Fig.1E). The lead compound has pharmaceutical range efficacy in the cell-based assays with GI50 ~ 0.9 µM. The therapeutic index and p53-independent findings are consistent also with our previous pre-clinical and clinical observations using decitabine or 5-azacytidine to inhibit the corepressor DNMT1 in AML cells (reviewed in Velcheti et al., ASCO Ed. Book 2017). Drug pharmacology and pre-clinical in vivo proof-of-principle experiments using patient-derived xenotransplant models of leukemia are in progress. Thus, druggable corepressors are the barriers between AML (and other cancer) cells and the terminal lineage-fates intended by their master transcription factor content, opening the door to novel non-cytotoxic, normal hematopoiesis sparing, differentiation-based oncotherapy.
Saunthararajah:Novo Nordisk, A/S: Patents & Royalties; EpiDestiny, LLC: Patents & Royalties.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal